DFF supports the point charge model of electrostatic interaction representation. Although it has its limitations, this simple model is known to be capable of predicting many properties accurately. A good example is liquid methanol, a system containing strongly polarized and hydrogen bonded molecules. Using atomic point charges derived from ab initio electrostatic potentials in a force field coupled with optimized van der Waals and valence terms works very well.
Charge parameters, either atom-type based or bond-type based, can be derived by fitting ab initio electrostatic potentials (ESP).[1] In fitting the electrostatic potentials, certain restrictions might be applied. This is necessary either because of shared (transferred) charge parameters or because of the difficulties in determining charges for atoms shielded by others in bulky molecules.[2, 3]
The following chart shows how the charge parameters are derived.
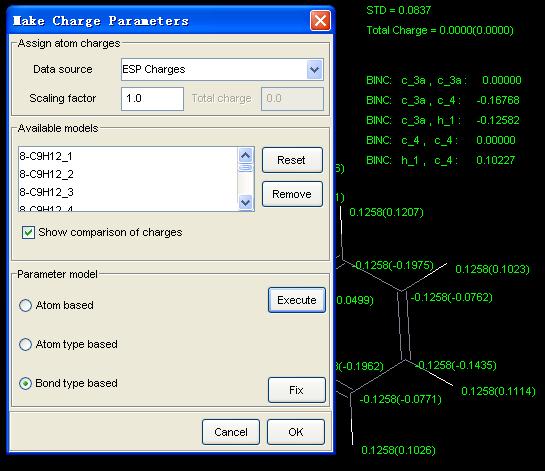
[1] Williams, D. E., J. Comp. Chem., 1988, 9, 745.
[2] C.I. Bayly, P. Cieplak, W.D. Cornell, and P.A. Kollman, J. Phys. Chem. 1993, 97, 10269.
[3] H. Sun, J. Phys. Chem. 1998, B102, , 7338-7364, .